导读
深海冷泉含有丰富多样的还原脱卤酶编码基因(rdhA),但其还原脱卤的潜力在很大程度上尚未开发。研究了冷泉沉积物微宇宙中2,4,6-三溴苯酚(TBP)的微生物脱溴作用。通过对不同营养源和底物浓度的培养条件进行优化,建立了一株高效脱溴微生物菌群,该菌群能够在72 h内完全降解50 μM TBP。宏基因组分析显示Bin 3是一种新的细菌,隶属于消化球菌科,是一种关键的脱卤剂,携带多个rdhA基因。微生物群落分析表明,养分的有效性显着影响β多样性(社区组成),但只有轻微的影响α多样性。通过降解动力学、共现网络分析、归一化随机性比分析和宏基因组定量,我们发现补充乳酸沿着0.05%的酵母提取物显著提高了TBP降解效率,并促进了关键脱卤微生物的靶向富集(相对丰度从<1%增加到32%)。比较基因组分析表明,Bin 3通过扩展参与皮利形成、细胞运动、营养获取和多种代谢途径的基因家族而经历了特定的适应,潜在地增强了其在深海冷泉环境中的竞争力。这项研究推进了我们对深海脱卤微生物及其对极端环境的适应性的理解,为其在污染物生物修复中的生态意义和潜在应用提供了见解。
论文ID
原名:Harnessing deep-sea cold seep microbiomes for reductive dehalogenation: from culturomics and genomics insights
译名:利用深海底冷泉微生物组进行还原脱卤:从培养组学与基因组学视角解析
期刊:Water Research
IF:12.4
发表时间:2025.6
DOI号:10.1016/j.watres.2025.124072
结果
养分变化对还原性脱溴活性的影响
接种TBP-10后,所有营养改良的培养体系都表现出微生物还原脱溴活性,而非生物对照则没有降解。 在被试组中,LY组的脱溴效率最高,在7天内将TBP完全脱卤为苯酚。 相反,LA组表现出最低的效率,需要21天才能去除TBP, 4BP(78.9%)是主要产物。 这表明,在一定的实验条件下,添加0.05%酵母提取物显著提高了所测试的冷渗微生物的还原脱溴,提高了效率和降解程度(图1)。 基于降解产物的顺序外观和浓度变化,微生物tbp脱溴优先针对邻位,其次是对位。
不同营养物培养组的微生物群落
通过16S rRNA基因扩增子测序,获得1.5 Gb的序列数据(平均读取长度为373 bp),得到5485个扩增子序列变异(asv)。 Alpha多样性指标(Shannon、Simpson和Chao1指数)显示,不同类群之间的物种多样性、丰富度或均匀性没有显著差异。 相比之下,beta多样性分析显示各组之间存在显著的组成差异。 维恩图进一步支持了这一发现,显示所有文化中只有44种共有的asv,而每个群体都有大量独特的asv,从659到1122不等。 为了阐明潜在的装配机制,我们采用了NST分析,该分析为评估这些过程提供了定量框架,阈值为50%作为更确定性(< 50%)和更随机(bbb50 %)之间的边界点。 结果显示了不同的生态过程:LY和BU组都表现出主要的确定性组装(NST < 30%),而其他组则表现出更多的随机影响。 这些发现共同表明:(1)营养条件显著重塑微生物群落组成,但不影响总体多样性;(2)不同培养条件下的群落组装机制存在很大差异。
为了更详细地表征微生物群落组成,我们对所有实验组中最丰富的前10个分类群的相对丰度进行了研究。 在所有营养条件下,杆菌门和假单胞菌门始终是两个最占优势的门,主要以梭状芽孢杆菌(27.5 - 82.2%)和γ蛋白细菌(4.5 - 45.5%)类为代表。检测到目前公认的相对丰度超过0.5%的还原性脱卤微生物。 值得注意的是,硫酸盐还原菌(SRB) Desulfotomaculum存在于这些培养物中,先前的研究报道了许多SRB含有rdhA基因。 为了评估这种可能性,我们对NCBI中目前可用的两个具有代表性的Desulfotomaculum基因组进行了基因组筛选(登录号:GCA_000214435和GCA_001655685)。 然而,全面分析未能在这些基因组中确定任何rdhA同源物,这表明负责观察到的脱卤活性的关键微生物仍有待确定。 LEfSe分析在除LA外的所有培养物中发现了群体特异性生物标志物(LDA bbb2.0),这些培养物与亲代TBP-10接种物保持相同的条件,进一步反映了营养驱动的群落变化。 LY组与盐杆菌科表现出特别强的相关性,这一点得到了共存网络的证实,其中Bacillota主导了相互作用。 这些嗜盐厌氧菌可以代谢三甲胺和甜菜碱等降解中间体。 网络分析进一步发现,BU组具有最高的复杂性(节点/边),而AC组具有随机的网络结构和关联(聚类系数= 0.081)。 鉴于高降解效率、确定性组装和适度的群落复杂性,LY群代表了识别关键脱卤微生物的有希望的目标。
不同盐度和TBP浓度对还原脱溴的影响
LY微生物联合体在广泛的盐度范围内表现出强大的TBP降解能力,在所有测试条件下,在五天内完全转化为苯酚。 当暴露于不同的TBP浓度时,该联合体表现出浓度依赖的降解动力学。 在TBP≤50 μM时,3天内完全降解为苯酚,平均溴化物(Br⁻)的去除率为1.5 μM d。 在150 μM TBP的前3天,观察到最高的Br -毒血症(4 μM d毒血症)。 相反,在200 μM的TBP下,Br -⁻的去除效率显著下降(Pttest < 0.01),最初3天的Br -⁻去除率下降到0.3 μ d。 这些结果表明,海洋来源的LY联合体在低盐度条件下保持强大的TBP降解活性,而其脱溴动力学表现出浓度依赖的动力学。 尽管需要延长孵育期(9天),但该团队最终在200 μM下实现了TBP的完全降解。
LY微生物联合体中关键还原脱卤细菌及其相关rdhA基因
基于宏基因组组装和随后的分组分析,共获得13个高质量的mag(完整性≥50%,污染≤10%)。 根据基因鉴定标准,在其中两个MAGs中鉴定出6个rdhA基因,其中Bin3中检测到5个,Bin9中检测到1个。 GTDB数据库分类显示,Bin3属于Peptococcia纲(GTDB分类组),而Bin9属于Oxobacteraceae科。 对来自Bin3的1016 bp的16S rRNA基因片段进行进一步分析发现,当与- microaceticum菌株DMC的二氯甲烷降解菌Dehalobacterium的16S rRNA基因序列比对时,该菌株的总分最高(覆盖率= 100%,一致性= 90.56%)。 根据已建立的分类阈值(属为94.5%,科为86.5%), Bin3可能隶属于胃球菌科,但与去盐杆菌属不同。 CoverM定量分析结果显示,群落中Bin3和Bin9的相对丰度分别达到32.2%和7.7%。 此外,通过使用本地BLAST将Bin3的16S rRNA基因序列与亲本代的代表性16S rRNA基因序列比对,我们确定Bin3在亲本培养孵育结束时的相对丰度低于1%。 这表明Bin3发生了显著的富集。6个rdhA基因与功能表征的rdhA基因的同源性为28.8% ~ 40.1% ,这表明它们可能代表了新的rdha。 重建的系统发育树进一步支持了这一假设,其中几个基因形成了独立的分支,如bin3_rdhA1和bin9_rdhA6。 系统发育分析表明,Bin3中的rdhA基因可能具有不同的进化起源,因为它们与来自不同脱卤微生物的rdhA序列聚集在一起。 此外,对含有这些rdhA基因的基因组背景分析表明,除了与膜和核苷酸亚基RdhB共编码外,它们还经常与电子传递复合物和转录调节因子相关。
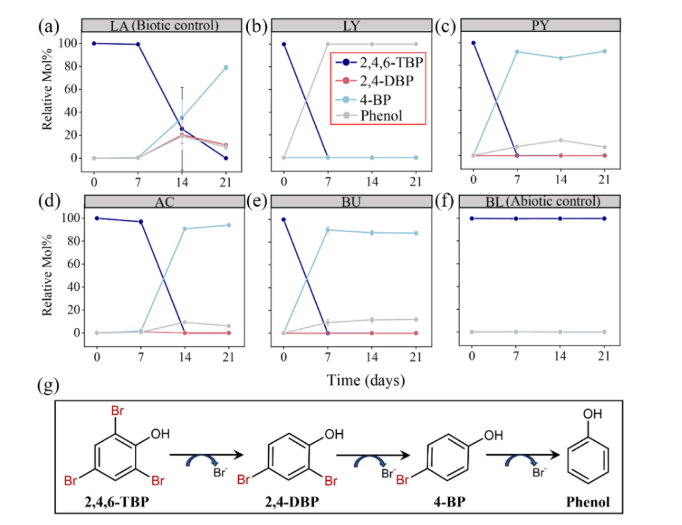
图1所示。 不同营养物对微生物还原脱溴的影响。 (a)乳酸(作为生物防治,LA); (b)乳酸(添加0.05%酵母提取物,LY); (c):丙酮酸酯(PY); (d):醋酸(加5%氢气,AC); (e):丁酸酯(BU); (f):非生物控制(BL); (g):还原脱卤途径。 图(a-f)显示了TBP及其脱溴产物的相对摩尔百分比(Mol %)的时间变化。 误差条表示标准差(n = 3)。
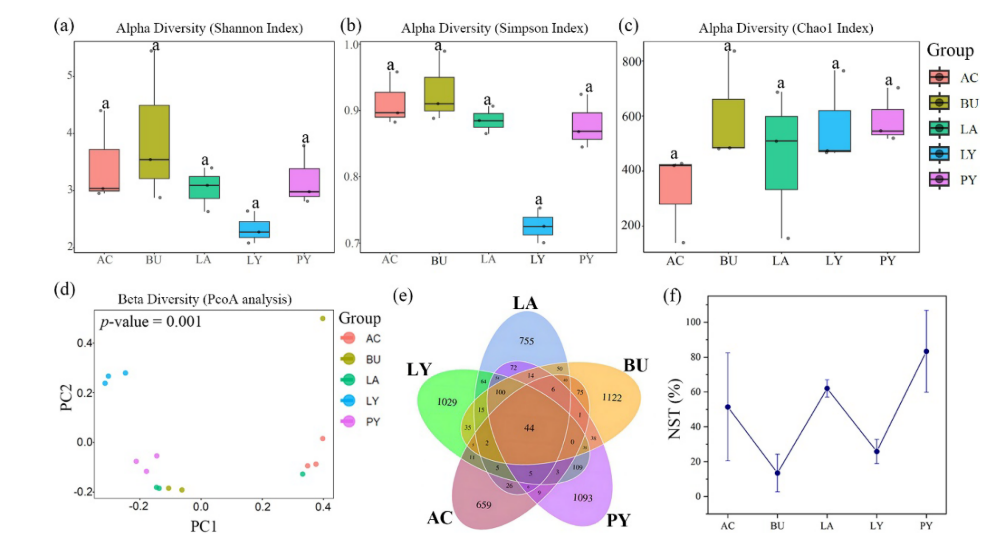
图2所示。 微生物群落多样性分析及群落组装评价。 (a-c): Alpha多样性分析; (d): Beta多样性分析; (e):维恩图解; (f): NST分析。 误差条表示标准差(n = 3)。
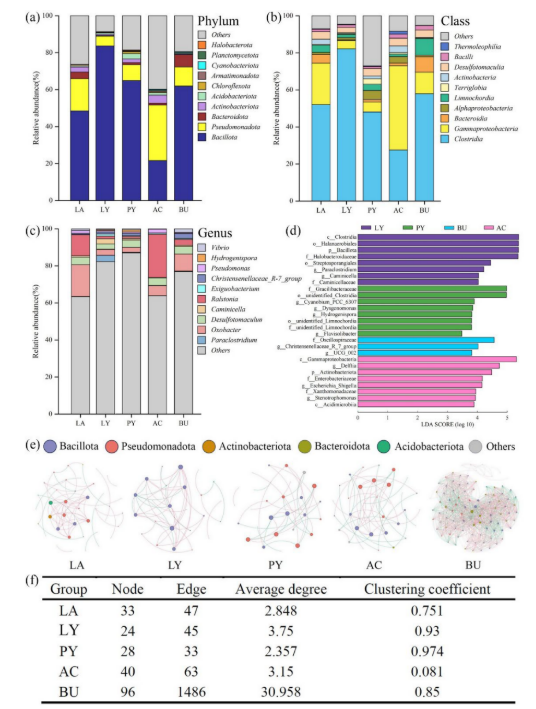
图3所示。 微生物群落结构,生物标志物和微生物共发生网络在不同的培养组。 (a-c):微生物群落组成; (d): LEfSe分析; (e-f):微生物共生网络和网络的拓扑特性。
结论
这项研究强调了深海冷泉微生物组在持久性溴化污染物还原脱卤方面尚未开发的潜力,利用了文化组学,宏基因组学和比较基因组学的组合。本工作的主要贡献包括建立了一个能够在不同NaCl浓度或TBP浓度(200 μM)下有效脱溴的培养体系。此外,我们还深入了解营养驱动的群落组装在富集脱卤微生物方面的作用。此外,我们确定了一种新的消化球菌科细菌作为TBP降解的关键球员。多个rdhA基因的存在,沿着与皮利形成、运动性、营养获取和多种代谢途径有关的基因家族的扩展,表明该细菌经历了环境适应性进化,可能增强其在深海冷泉中的存活。此外,其基因组种间直接电子转移的潜力提出了有趣的问题,其可能的代谢与厌氧甲烷古菌的相互作用。总的来说,这项工作弥合了海洋微生物生态学和应用生物修复之间的关键差距,扩大了海洋脱卤剂的基因组数据集,提高了对其进化和功能多样性的理解。
